La enfermedad de Huntington es una enfermedad neurodegenerativa hereditaria y letal producida por una mutación en el gen denominado huntingtina (HTT). Mutaciones en HTT afectan gravemente al cerebro, llevando a muerte neuronal severa en el estriado y en otras zonas del cerebro, como la corteza, en menor medida. Los pacientes con enfermedad de Huntington presentan síntomas motores, psiquiátricos y cognitivos que avanzan progresivamente hasta la muerte, la cual ocurre aproximadamente 20 años después del diagnóstico. No existe tratamiento disponible para estos pacientes.
A pesar de que la causa genética de la enfermedad de Huntington se conoce, las consecuencias biológicas de esta mutación todavía no se comprenden por completo. La huntingtina mutante se expresa en neuronas pero también en células no neuronales como los astrocitos. Los astrocitos están en íntimo contacto con las neuronas y realizan una amplia variedad de funciones que incluyen el mantenimiento de la homeóstasis neuronal, la regulación de la señalización neuronal y la respuesta a daño cerebral. Se ha observado que algunas de estas funciones se alteran en la enfermedad de Huntington. Sin embargo, numerosas cuestiones surgen a partir de los estudios previos (Khakh BS et al, 2017): ¿Cuál es sustrato molecular para el desarrollo de estos fenotipos en astrocitos? ¿Qué otras alteraciones se producen en estas células? ¿Es la alteración de los astrocitos fruto de mecanismos autónomos del astrocito o depende de los cambios en otras células?
En el laboratorio del Dr. Baljit Khakh nos propusimos arrojar un poco de luz sobre estas cuestiones mediante el análisis detallado de los cambios que se producen en la expresión génica y proteica de los astrocitos en muestras de pacientes humanos y en modelos de ratón para la enfermedad de Huntington.
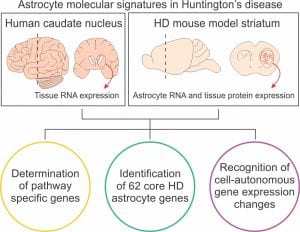
En un artículo, recientemente publicado en Science Translational Medicine, realizamos secuenciación de ARN de astrocitos del estriado de dos modelos de ratón con enfermedad de Huntington a tres estadios diferentes de la enfermedad: presintomática, sintomática y estadio muy avanzado. Los resultados fueron comparados con los transcriptomas de tejido completo de pacientes con enfermedad de Huntington y proteomas de ratón de la misma región cerebral, en estadios de la enfermedad equivalentes. El análisis detallado de los datos identificó los principales cambios de expresión génica en astrocitos de enfermedad de Huntington a lo largo de la progresión de la enfermedad. Utilizamos dicha información para responder las cuestiones planteadas previamente.
¿Cuál es el sustrato molecular para el desarrollo de fenotipos de enfermedad de Huntington en astrocitos?
Los cambios funcionales de los astrocitos en la enfermedad de Huntington descritos en estudios previos incluyen alteraciones en la señalización de calcio, la absorción de neurotransmisores, la regulación de la concentración de potasio, el metabolismo y la adquisición de un fenotipo reactivo relacionado con la neuroinflamación.
Encontramos expresión diferencial de genes implicados en el transporte de calcio, potasio y neurotransmisores que podría estar relacionada con las observaciones previas. Además, detectamos alteraciones en la expresión de genes relacionados con el metabolismo, incluyendo una disminución muy acusada en la síntesis de colesterol, así como de múltiples genes implicados en morfología y adhesión celular.
Inesperadamente, aunque se ha propuesto que la reactividad de los astrocitos podría estar causando muerte neuronal en las enfermedades neurodegenerativas, incluyendo la enfermedad de Huntington (Liddlelow SA et al, 2017), la exploración detallada de nuestros datos y los análisis de expresión en humanos no mostraron evidencias de reactividad de los astrocitos hasta los últimos estadios, más graves, de la enfermedad.
¿Qué otras alteraciones no descritas experimentan los astrocitos?
En general, el número de genes cuya expresión se altera progresa con la severidad de la enfermedad. No obstante, detectamos un solapamiento amplio de genes entre los dos modelos de ratón y los tres estadios de la enfermedad estudiados. Para algunos de estos genes, la magnitud del cambio aumentaba gradualmente con la progresión de la enfermedad. El análisis detallado de los datos reveló nuevas rutas moleculares interesantes para estudios futuros como, por ejemplo, la señalización mediante el receptor asociado a proteína G (GPCR, del inglés G protein coupled receptor), cAMP o Wnt.
Identificamos una huella de expresión génica de los astrocitos en la enfermedad de Huntington mediante la comparación de los genes de astrocitos con expresión alterada en los dos modelos de ratón con los cambios de expresión de genes en humanos y de proteínas de ratón de la misma región cerebral. Consideramos esta huella como una representación de los cambios primordiales que ocurren en los astrocitos en la enfermedad de Huntington. La huella incluye 62 genes implicados en funciones como la señalización mediada por GPCR, la señalización de calcio, la absorción de neurotransmisores y comportamientos dependientes del estriado.
¿Son las alteraciones funcionales de los astrocitos provocadas por mecanismos autónomos del astrocito o dependen de los cambios en otras células?
Para saber si las alteraciones previamente descritas se deben a los efectos de la expresión de la huntingtina en los astrocitos (mecanismos celulares autónomos) o por el contrario, son consecuencia de los cambios ocurridos en el circuito que los rodea (no autónomos), expresamos una proteína con dedos de zinc (ZFP, del inglés zinc finger protein) en astrocitos para reprimir de forma específica la expresión de la huntingtina mutante en estas células. La expresión de ZFP en astrocitos redujo los niveles de huntingtina mutante al 30% de la cantidad acumulada en los astrocitos que no expresaban ZFP. Los análisis de secuenciación de ARN de los modelos de ratón de enfermedad de Huntington que expresaban ZFP demostraron que la reducción de huntingtina mutante en astrocitos redujo la magnitud de los cambios de expresión génica de 61 de los 62 genes de la huella de expresión génica de astrocitos en la enfermedad de Huntington.
Además, encontramos que las rutas moleculares relacionadas con la enfermedad de Huntington, el daño sobre el ADN y la señalización mediada por Wnt se restauraban. Finalmente, a partir de estos experimentos identificamos a Adora2a, un GPCR capaz de regular algunos de los 62 genes de la huella molecular. La expresión de Adora2a también está alterada en los astrocitos de enfermedad de Huntington y se recupera mediante la expresión de ZFP.
En resumen, nuestros resultados definen los cambios de expresión génica en los astrocitos en enfermedad de Huntington, identifican los efectos celulares autónomos de la expresión de HTT mutante en astrocitos y desvelan rutas de señalización en estas células, como la señalización a través de GPCR, que podrían ser explotadas como dianas de tratamiento.
Blanca Diaz-Castro, UK Dementia Research Institute at The University of Edinburgh.
Fuente: http://bit.ly/2OVjqFV




