
En los últimos 15 años, los estudios de asociación de genoma completo (GWAS por sus siglas en inglés) han sido claves para la identificación de variantes genéticas asociadas a rasgos o enfermedades. Los llamados “hits” de GWAS aparecen en clústeres de variantes que se encuentran en desequilibrio de ligamiento. Sin embargo, estos estudios se centran en la variante de nucleótido único (SNV por sus siglas en inglés) principal, que es la más significativa, mientras las SNVs que la rodean, que presentan asociaciones más bajas, son consideradas redundantes.
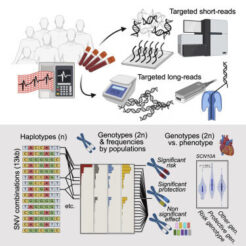
En este trabajo, Pinsach-Abuin y colaboradores estudiaron la importancia de integrar el conjunto hits de GWAS de un clúster—en lugar de estudiar variantes genéticas individuales—para mejorar la información generada por los datos de GWAS. Para realizar este estudio, se centraron en el síndrome de Brugada, un trastorno eléctrico cardíaco hereditario caracterizado por arritmias ventriculares y riesgo de muerte súbita cardíaca. La mayor parte de las variantes genéticas deletéreas asociadas a este síndrome se localizan en el gen SCN5A y, en menor proporción, en el gen SCN10A, que codifican para los canales de sodio regulados por voltaje NaV1.5 y NaV1.8, respectivamente. Sin embargo, estas variantes patogénicas corresponden sólo al 35% de los casos diagnosticados, con lo que aún existe un 65% de pacientes con síndrome de Brugada en los que el defecto genético es desconocido.
En base a estos antecedentes, y teniendo en cuenta que el locus SCN5A–SCN10A contiene numerosos hits de GWAS asociados alteraciones de conducción cardíaca o del electrocardiograma, los investigadores estudiaron la posibilidad que el origen genético del síndrome de Brugada pudiera ser atribuido, en parte, a grupos de variantes en forma de haplotipos en este locus.
En primer lugar, caracterizaron la estructura del locus SCN5A-SCN10A, identificando dos bloques de haplotipos que acumulan la mayoría de los SNV comunes asociados al síndrome de Brugada, a los que denominaron bloques promotor y enhancer, respectivamente. A continuación, analizaron los SNVs de esta región en 86 pacientes con síndrome de Brugada mediante secuenciación de última generación de lectura corta y larga (short-read and long-read sequencing), y los compararon con datos genómicos de 3 grupos control de ascendencia europea. Este análisis reveló la presencia de cinco haplotipos comunes en la población europea (Hap1-5), formados por la combinación de 7 SNVs comunes en el bloque denominado enhancer.
Curiosamente, aunque los Hap1-3 aparecen asociados al síndrome de Brugada, más de la mitad de los casos estudiados eran homocigotos para Hap1 (Hap1/1), mientras que ningún caso presentaba el genotipo Hap2/3. De acuerdo con esta observación, los análisis de odds ratio identifican a Hap1/1 como haplotipo de riesgo al síndrome de Brugada, y a Hap2/3 como genotipo protector.

Teniendo en cuenta que Hap1 y Hap2 comparten cuatro alelos (rs2, rs3, rs5 y rs7), estos resultados demuestran que los efectos de riesgo y protección dependen del contexto del haplotipo y del genotipo. La relevancia de este estudio en relación al síndrome de Brugada está en la observación que Hap1, el haplotipo de riesgo, se ajuste a un modelo de herencia recesiva, así como en la identificación de un genotipo protector al síndrome en una región no codificante.
En conclusión, el trabajo de Pinsach-Abuin y colaboradores demuestra la importancia de estudiar las variantes genéticas asociadas a enfermedad como haplotipos, ya que cuando se estudian de forma individual se puede perder información genética relevante. Este estudio representa un nuevo enfoque en este tipo de análisis genéticos, y propone reanalizar los datos de GWAS previamente publicados.
Mel·lina Pinsach-Abuin1,2,3, Ivan Garcia-Bassets4 y Sara Pagans1,2,3
1 Department of Medical Sciences, School of Medicine, Universitat de Girona, Girona, Spain
2 Institut d’Investigació Biomèdica de Girona, Salt, Spain
3 Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares, Madrid, Spain
4 Department of Medicine, School of Medicine, University of California, San Diego, La Jolla, CA, USA
Fuente: https://genotipia.com/genetica_medica_news/genetica-sindrome-de-brugada/




