
La enfermedad de Alexander es una leucodistrofia que produce una alteración en la constitución de la mielina. El mecanismo de ganancia de función de la proteína ácida fibrilar glial es una de las hipótesis que existe sobre sus causas.
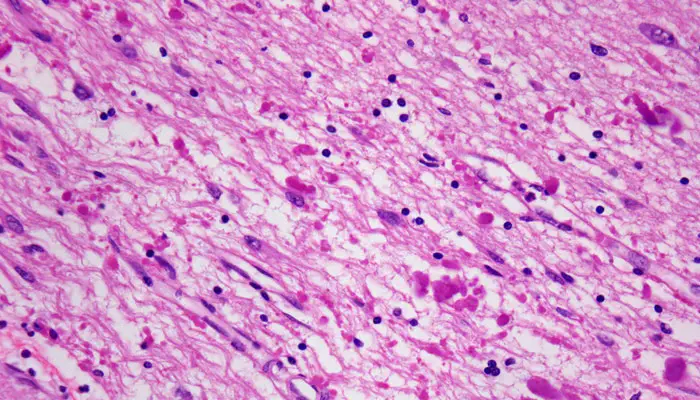
Las leucodistrofias son un grupo de enfermedades hereditarias que afectan la sustancia blanca del sistema nervioso central (SNC) y ocasionalmente del sistema nervioso periférico.
La enfermedad de Alexander (AxD) es una de ellas y su base patológica, junto con la pérdida de mielina, es la aparición de los cuerpos de Rosenthal (inclusiones citoplasmáticas en las células gliales descritas también en determinados gliomas).
Estos depósitos están constituidos por la proteína ácida fibrilar glial (PAFG) y otros componentes como la vimentina y la plectina (ambas proteínas de unión al citoesqueleto).
Desde el punto de vista clínico, la enfermedad tiene formas: infantil, juvenil y adulta, siendo la primera la de peor pronóstico.
Las mutaciones en el gen que codifica la PAFG se han identificado como la base genética de la AxD. Estas mutaciones consisten en cambios en 32 nucleótidos específicos y aparecen tanto en casos familiares como esporádicos.
Sin embargo, no se conoce cómo estas variantes conducen a la agregación de la PAFG, ni el mecanismo que relaciona a esta proteína con la pérdida de mielina.
Lo que sí es un hecho es que la desmielinización forma parte del cuadro clínico y que en los casos de mayor supervivencia llega a ser muy intensa, afectando prácticamente toda la sustancia blanca.
Características de la PAFG
Esta proteína es un componente de los filamentos intermedios de los astrocitos.
Estos filamentos, aunque contribuyen a la estructura celular y forman el citoesqueleto junto con los microtúbulos y los microfilamentos, tienen una función importante en la transmisión de señales.
La PAFG también está presente en otras células como las de Schwann (sistema nervioso periférico) y las células de la glía entérica (sistema nervioso entérico).
Hipótesis 1: Ganancia de función de la PAFG

Quienes defienden esta teoría argumentan que la enfermedad se produce por el acúmulo de PAFG en los astrocitos y que la elevación de los niveles de esta proteína es dañina.
Por lo que han planteado un mecanismo de ganancia de función, similar al que se ha propuesto en otras enfermedades neurodegenerativas, como en la esclerosis lateral amiotrófica causada por SOD1.
El mecanismo que explica esta hipótesis está en discusión, no obstante, lo analizaremos a continuación:
- La PAFG se agrega secuestrando a la vimentina, plectina y proteínas de choque térmico.
- Posteriormente ocurre la fosforilización y la unión de otros elementos proteicos, lo cual genera los cuerpos de Rosenthal.
- Esta situación desencadena la lesión astrocitaria, a la vez que inicia la activación de vías de respuesta al estrés celular.
Esta hipótesis ha generado un número importante de experimentos y modelos, especialmente en ratones que sobreexpresan la PAFG tanto mutada como normal.
Asimismo, se ha tratado de correlacionar el pronóstico de la enfermedad con la determinación de PAFG en el líquido cefalorraquídeo.
Con ello se ha conseguido una importante información sobre qué mecanismos se desarrollan a consecuencia del acúmulo de PAFG.
¿Por qué está en discusión esta hipótesis sobre el origen de la enfermedad de Alexander?
- El principal problema de esta hipótesis es que no explica la forma en que se genera la desmielinización.
- Otra cuestión que limita esta hipótesis es que en los gliomas que presentan acúmulo de PAFG y que desarrollan cuerpos de Rosenthal tampoco hay perdida mielínica.
Es importante señalar que no se trata de negar los efectos negativos de los niveles elevados de PAFG, sino que en base a esas limitaciones es muy probable que este mecanismo no explique por sí solo las causas de la enfermedad.
Lechner Rodriguez Aguilar
Artículos relacionados: leucodistrofias, enfermedades hereditarias, mielina,
Fuente: http://bit.ly/2Cq3t46




