Sinónimos: –
Prevalencia: Desconocido
Herencia: Autosómico recesivo
Edad de inicio o aparición: Infancia / Neonatal
Resumen
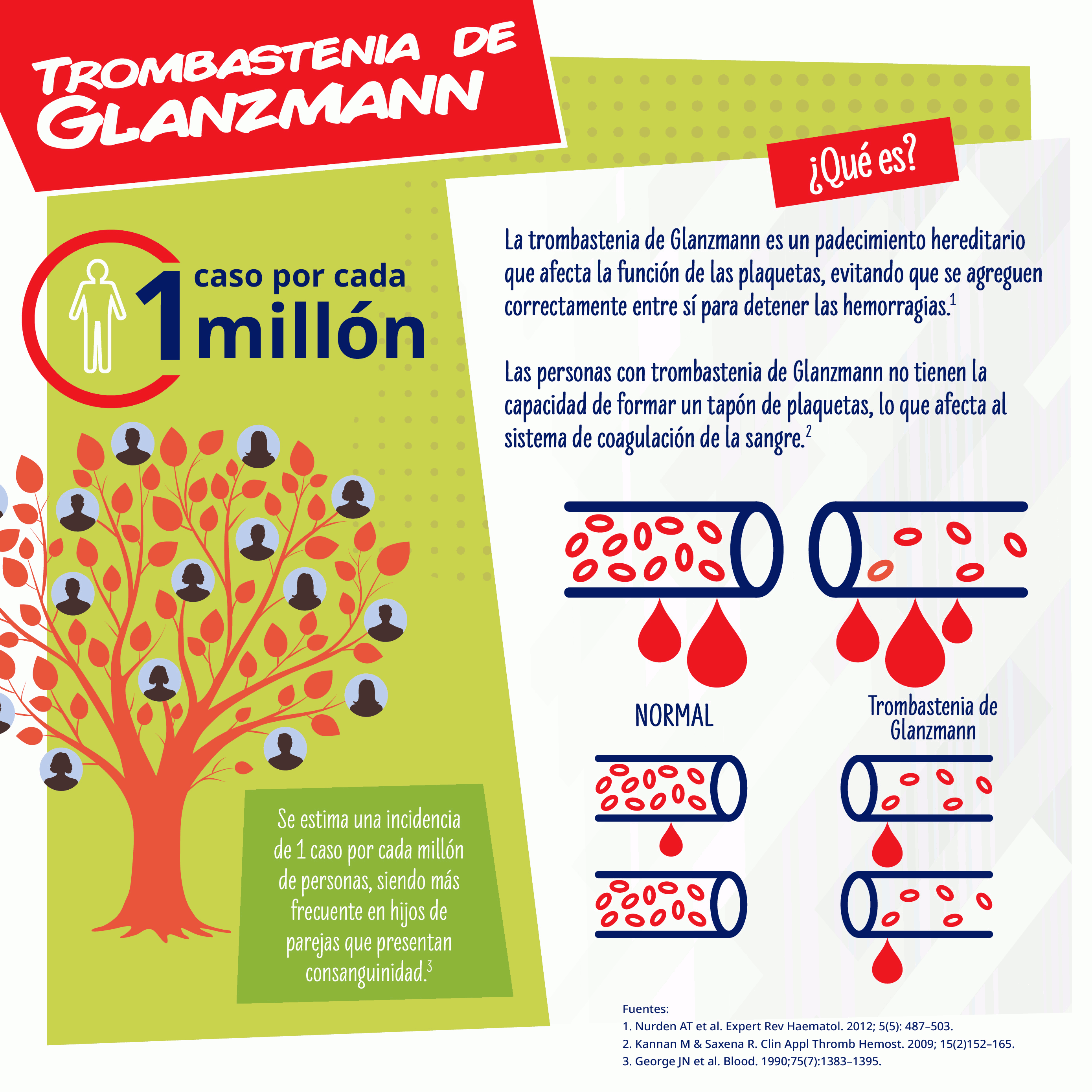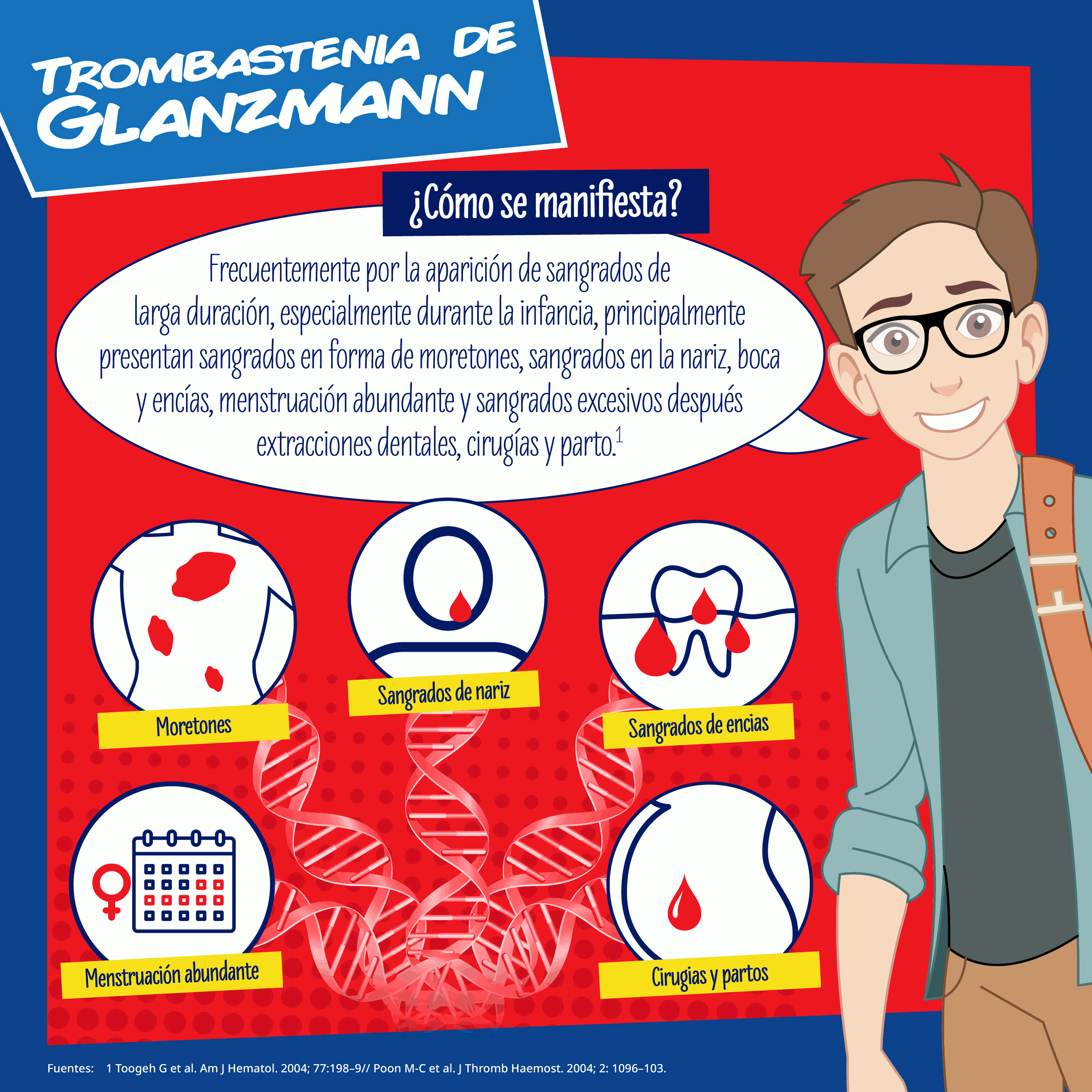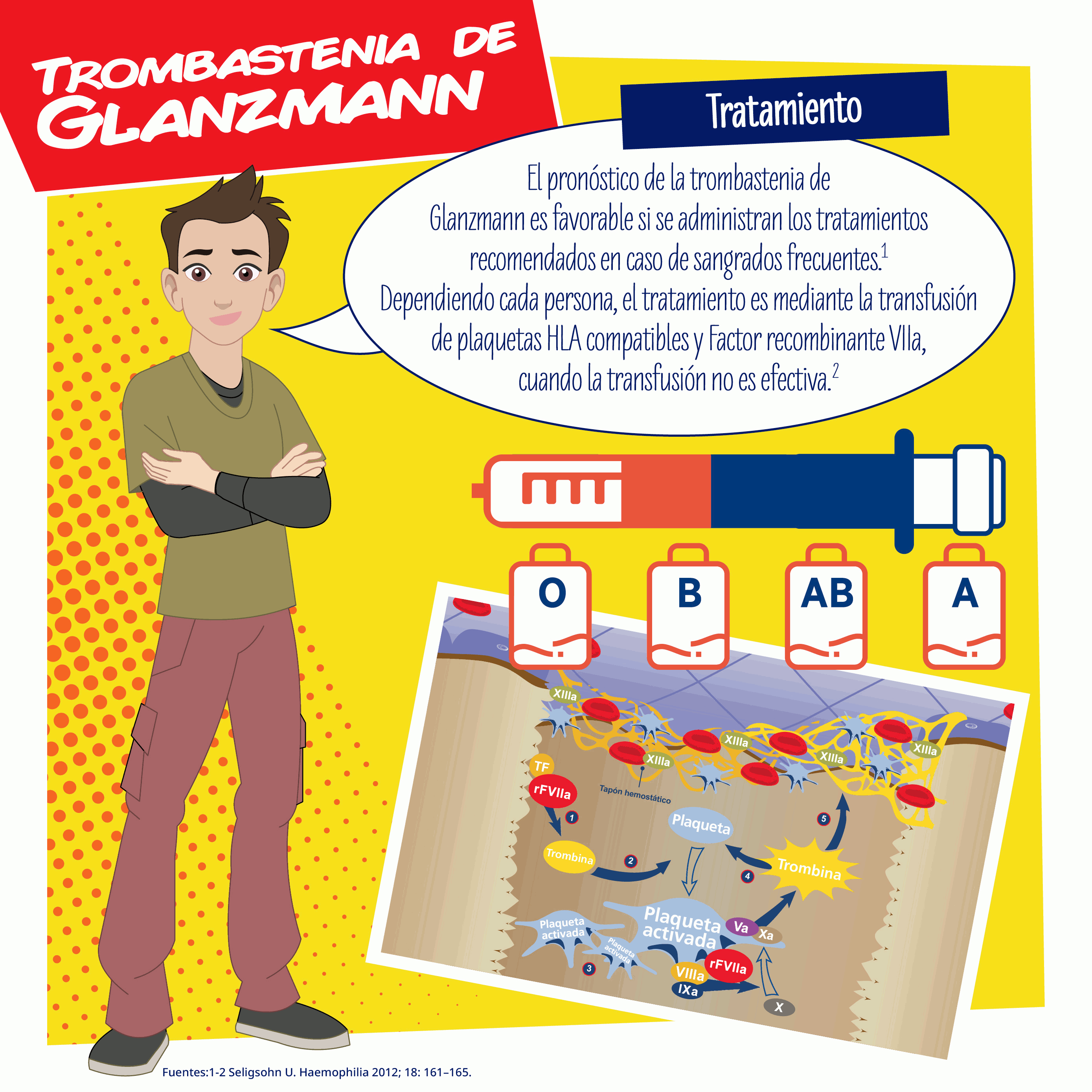
La trombastenia de Glanzmann (GT, por sus siglas en inglés) es un padecimiento hereditario de la coagulación que afecta al linaje megacariocítico y que se caracteriza por la ausencia de agregación plaquetaria. Afecta la función de las plaquetas, evitando que se agreguen correctamente entre sí para detener las hemorragias. Las personas con esta enfermedad rara no tienen la capacidad de formar un tapón de plaquetas, lo que afecta al sistema de coagulación de la sangre.
A pesar de desconocerse la prevalencia, este síndrome es poco frecuente. Se estima una incidencia de 1 caso por cada millón de personas, siendo más frecuente en hijos de parejas que presentan consanguinidad.
La GT presenta una gran variabilidad clínica: algunos pacientes solamente tienen tendencia a presentar hematomas con contusiones mínimas, mientras que otros tienen hemorragias frecuentes, graves y potencialmente fatales. El lugar de sangrado en la GT está bien definido: púrpura, epistaxis, hemorragia gingival, y menorragia son características prácticamente constantes; la hemorragia gastrointestinal y la hematuria son menos comunes. En la mayoría de los casos, los síntomas de sangrado se manifiestan rápidamente después del nacimiento, a pesar de que la GT se diagnostique ocasionalmente en etapas posteriores.
El síndrome se transmite de forma autosómica recesiva.
La base molecular se asocia a anomalías cuantitativas y/o cualitativas de la integrina alphaIIb beta3. Este receptor media la unión de las proteínas de adhesión que unen los agregados plaquetarios y que asegura la formación del trombo en los vasos sanguíneos lesionados.
Se manifiesta clínicamente por sangrado de larga duración, especialmente durante la infancia cuando principalmente presentan hemorragias muco-cutáneas en forma de equimosis espontáneas (moretones), sangrado nasal, hemorragia en boca y encías, menstruación abundante y sangrados excesivos después extracciones dentales, cirugías y parto.
El diagnostico se basa en la asociación de sangrado mucocutáneo, ausencia de agregación plaquetaria en respuesta a todos los estímulos fisiológicos, y un recuento y morfología plaquetaria normales. La deficiencia o la no funcionalidad de la alphaIIb beta3 plaquetaria se debe confirmar siempre, mediante, por ejemplo, citometría de flujo.
Para evitar la allo-inmunización plaquetaria, el tratamiento terapéutico debe incluir, si es posible, procedimientos hemostáticos locales y/o la administración de DDAVP (desmopresina). Si estas medidas son ineficaces, o para evitar el sangrado durante una cirugía, se requiere con frecuencia una transfusión de concentrados plaquetarios HLA-compatibles. La administración del factor recombinante VIIa es otra alternativa terapéutica cada vez más usada.
La GT puede ser una enfermedad hemorrágica grave, sin embargo, el pronóstico es excelente con los tratamiento recomendados en caso de sangrados frecuentes.
Dependiendo cada persona, el tratamiento es mediante la transfusión de plaquetas HLA compatibles y factor recombinante VIIa, cuando la transfusión no es efectiva.
Revisores expertos
- Dr Alan NURDEN
Referencias y fuentes:
- Nurden AT et al. Expert Rev Haematol. 2012; 5(5): 487–503
- Kannan M & Saxena R. Clin Appl Thromb Hemost. 2009; 15(2)152–165
- George JN et al. Blood. 1990;75(7):1383–1395
- Toogeh G et al. Am J Hematol. 2004; 77:198–9// Poon M-C et al. J Thromb Haemost. 2004; 2: 1096–103
- Seligsohn U. Haemophilia 2012; 18: 161–165
- Trombastenia-de-Glanzmann_infografia
Última actualización: Abril 2006
Fuente: Orphanet (Trombastenia de Glanzmann)




